









Mukowiscydoza
W latach 2010-2012 NZOZ Genomed wykonywał badania genetyczne w ramach Programu Badań Przesiewowych Noworodków w kierunku mukowiscydozy na zlecenie Instytutu Matki i Dziecka. Program został zatwierdzony przez Ministerstwo Zdrowia w ramach polityki zdrowotnej w latach 2009-2014.
Poniżej przedstawiamy informacje dotyczące zarówno badań genetycznych wykonywanych w ramach Programu, jak również podstawowe informacje dotyczące samej choroby - mukowiscydozy.
W przypadku dodatkowych pytań i odpowiedzi, które chcieliby Państwo znaleźć na naszej stronie prosimy o kontakt e-mailowy:
diagnostyka@genomed.pl
lub telefoniczny: 22 6446019.
Serdecznie zapraszamy!!! ![]()
PODSTAWOWE INFORMACJE:
- Kiedy wykonywane jest genetyczne badanie przesiewowe noworodków w kierunku mukowiscydozy?
- Jaki jest zakres badań genetycznych w kierunku mukowiscydozy w Programie Przesiewowym w Polsce?
- Czy badanie genetyczne wykonywane w ramach programu przesiewowego noworodków w kierunku mukowiscydozy może wykluczyć lub potwierdzić chorobę u dziecka?
- Co to jest mukowiscydoza? Jak jest dziedziczona?
- Czy mutacje genu CFTR odpowiedzialne są tylko za mukowiscydozę?
- Co oznacza termin "dziedziczenie autosomalne recesywne"?
- Jaką funkcję pełni gen CFTR?
- Dlaczego diagnostyka genetyczna mukowiscydozy jest trudna?
- Dlaczego potrzebna jest porada genetyczna?
- Ośrodki zajmujące się diagnostyką kliniczną noworodków z nieprawidłowym wynikiem badania przesiewowego
![]()
Kiedy wykonywane jest genetyczne badanie przesiewowe noworodków w kierunku mukowiscydozy?
![]()
Jaki jest zakres badań genetycznych w kierunku mukowiscydozy w Programie Przesiewowym w Polsce?
| F508del | dele2,3(21kb) | 3849+10kbC>T | R334W |
| R347P | 1717-1G>A | G542X | G551D |
| R553X | 2184insA | 2143delT | 2183AA>G |
| K710X | 3272-26A>G | W1282X | N1303K |
Ponieważ do wykonania badania stosowana jest metoda sekwencjonowania - możliwe jest również wykrycie mutacji rzadko występujących w genie CFTR (<0.5%) lub mutacji nowych, wcześniej nie wykrytych u pacjentów z mukowiscydozą na świecie.
![]()
Czy badanie genetyczne wykonywane w ramach programu przesiewowego noworodków w kierunku mukowiscydozy może wykluczyć lub potwierdzić chorobę u dziecka?
![]()
Co to jest mukowiscydoza? Jak jest dziedziczona?
Mukowiscydoza (CF, ang. Cystic Fibrosis) jest chorobą genetyczną, powodowaną mutacjami (defektami, błędami) w obrębie genu CFTR. Jest jedną z najczęstszych chorób dziedziczonych w sposób autosomalny recesywny wśród rasy białej. Częstość CF w Europie wynosi 1/2500 żywo urodzonych, a w związku z tym nosicielem genu z defektem genetycznym jest co 25-ta osoba.
![]()
Czy mutacje genu CFTR odpowiedzialne są tylko za mukowiscydozę?
- Klasyczna postać CF z niewydolnoscią zewnątrzwydzielniczą trzustki
- Klasyczna postać CF z wydolnoscią zewnątrzwydzielniczą trzustki
- Atypowe postaci CF
- Noworodkowa hypertrypsynogenemia
- Izolowana postać azoospermii obstrukcyjnej
- Przewlekłe zapalenie trzustki
- Aspergillosa oskrzelowo-plucna
- Rozstrzenie oskrzeli
- Stwardniajace zapalenie dróg zółciowych
![]()
Co oznacza termin "dziedziczenie autosomalne recesywne"?
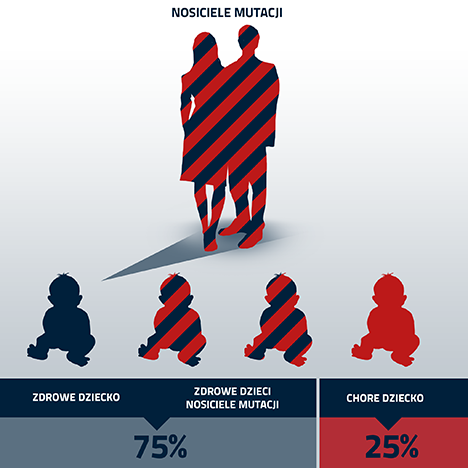
Para rodziców będących nosicielami, może mieć z prawdopodobieństwem 25% dziecko chorujące na CF. Prawdopodobieństwo to jest stałe i niezmienne przy każdych kolejnych ciążach. Należy również pamiętać, że istnieje 50% szansa, iż dziecko takiej pary będzie również nosicielem genu z defektem genetycznym, a tym samym będzie miało w przyszłości podwyższone ryzyko posiadania dziecka chorego na CF.
![]()
Dlaczego diagnostyka genetyczna mukowiscydozy jest trudna?
- wielkość genu
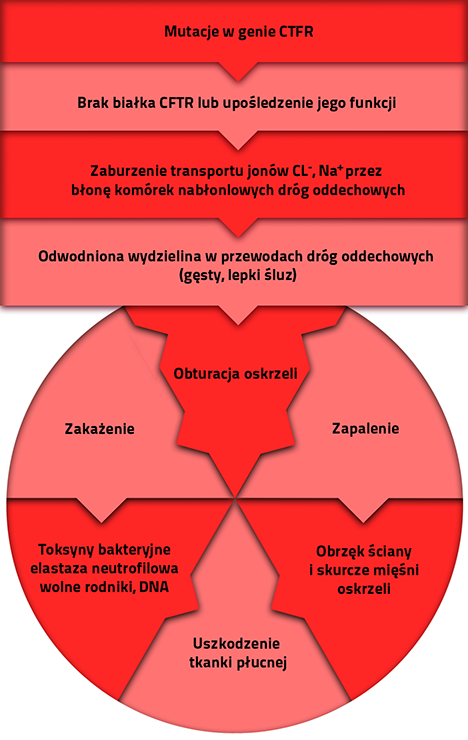
Gen CFTR jest jednym z największych genów człowieka (zbudowany jest z 310 000 par zasad!). Część tego obszaru zajmuje region kodujący białko, który składa się z 27 fragmentów (tzw. eksonów), pozostały obszar to regiony niekodujące - tzw. introny.
- liczbę mutacji
Obecnie znanych jest ponad 1600 mutacji genu CFTR. Dodatkowy problem stanowi fakt, iż wciąż pojawiają się doniesienia o identyfikacji nowych mutacji (defektów) u pacjentów z mukowiscydozą i niestety nie zawsze są to mutacje umożliwiające ich jednoznaczną interpretację i odpowiedź na pytanie dot. ich znaczenia dla funkcjonowania białka.
![]()
Dlaczego potrzebna jest porada genetyczna?
![]()
Ośrodki zajmujące się diagnostyką kliniczną noworodków z nieprawidłowym wynikiem badania przesiewowego :
- Dr Katarzyna Zybert
Klinika Pediatrii, Poradnia Mukowiscydozy i Chorób Płuc
Instytut Matki i Dziecka
ul. Kasprzaka 17A
01-211 Warszawa
tel: 22-327-7231
- Dr Maria Trawińska
Poradnia Mukowiscydozy
SZOZ nad Matką i Dzieckiem
ul. Polanki 119
80-308 Gdańsk
tel: 58-554-3165
- Prof. Halina Woś
Klinika Pediatrii
Śląski Uniwersytet Medyczny Górnośląskiego Centrum Zdrowia Dziecka
ul. Medyków 16
40-752 Katowice
tel: 32-207-1700
- Dr Zuzanna Kurtyka
Wojewódzki Specjalistyczny Szpital Dziecięcy im. Świętego Ludwika
ul.Strzelecka 2
31-503 Kraków
tel: 12-619-8634
- Dr Teresa Ruszczyk-Bilecka
Wojewódzki Szpital Specjalistyczny im. Mikołaja Kopernika
ul. Pabianicka 62
93-513 Łódź
tel: 42-6895875 (poradnia, środy)
tel: 42-689-5975 (oddział)
- Prof. dr hab. Jarosław Walkowiak
dr Aleksandra Lisowska
I Katedra Pediatrii
Klinika Gastroenterologii Dziecięcej i Chorób Metabolicznych
ul. Szpitalna 27/33
60-572 Poznań
tel: 61-849-1580
- Dr Iwona Ostrowska
Klinika Pediatrii, Endokrynologii, Diabetologii, Chorób Metabolicznych i Kardiologii Wieku Rozwojowego
Samodzielny Szpital Kliniczny nr 1 im. Prof. Tadeusza Sokołowskiego PAM
ul. Unii Lubelskiej 1
71-252 Szczecin
tel: 91-425-3168 (pielęgniarka)
tel: 91-425-3176 (pokój lekarski)
- Dr Janina Pawłowicz
Poradnia Alergologiczno-Pulmonologiczna
ul. Krasińskiego 29
50-450 Wrocław